
Andrew J Skelton
Data Science | Bioinformatics | Machine Learning | Statistics
Data Science | Bioinformatics | Machine Learning | Statistics





I work on clinical bioinformatics research in immunology and skeletal medicine. I'm passionate about data science and machine learning in a clinical research setting, maximising the valuable data we have.
I'm highly proficient in R, and love taking on new and complex data science problems. Take a look at my CV and projects, if you ever want a chat about a project, or job, then get in touch!


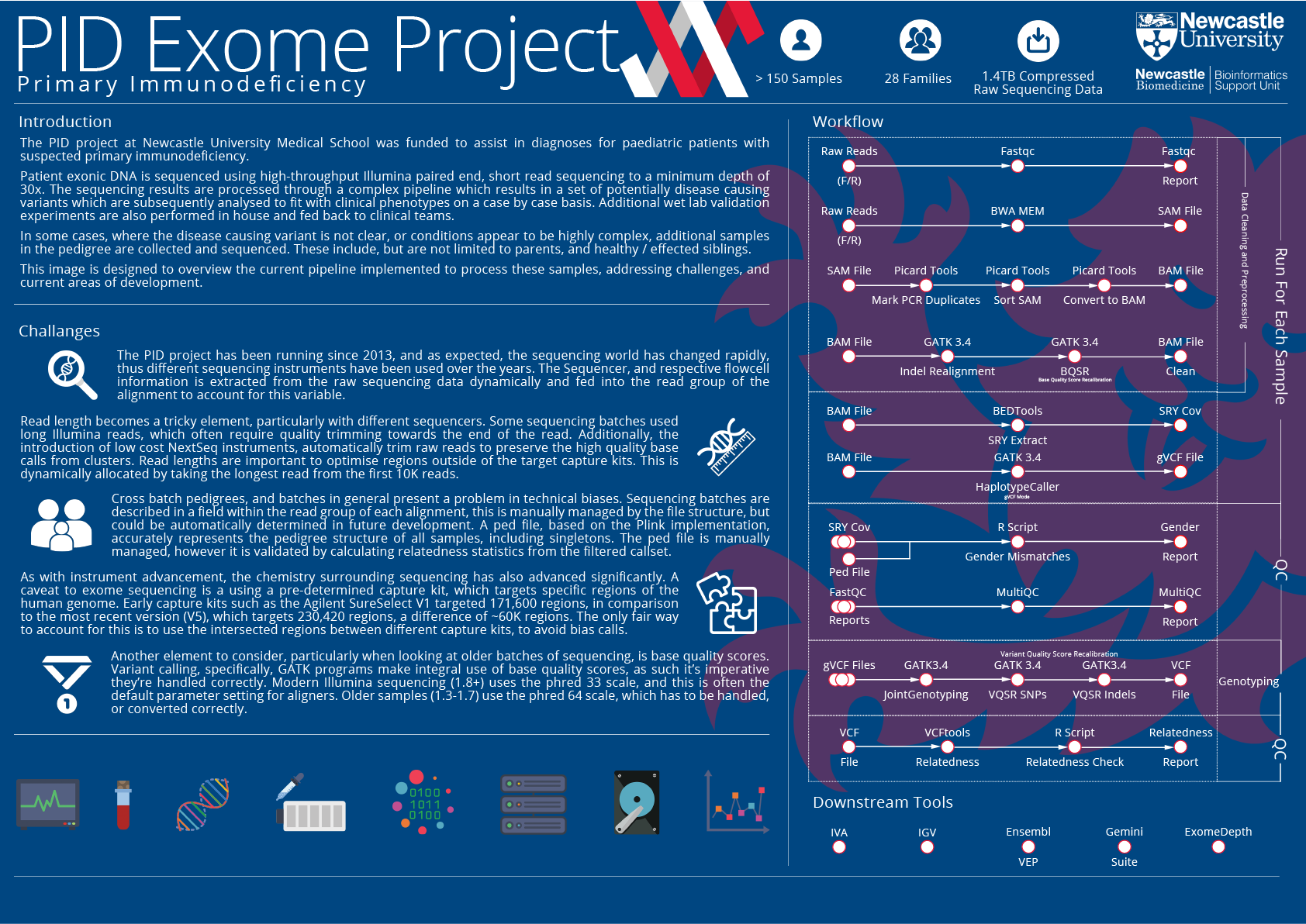

{kind=link}